



امروز جمعه ۲۱ اردیبهشت ۱۴۰۳
دسته بندی سایت
برچسب های مهم
پیوند ها
آمار بازدید سایت
درمان ژنتیکی بیماری پارکینسون، به روز رسانی

چکیده. درمان اصلی فعلی بیماری پارکینسون (PD)شامل درمان جایگزینی دوپامین است که علاوه بر ایجاد چندین عارضه جانبی، پیشرفت بیماری را به تاخیر نمیاندازد. حوزه ژندرمانی یک ابزار بالقوه برای بهبود درمان فعلی ارائه میدهد. بررسی حاضر، به روز رسانی وضعیت فعلی ژندرمانی برای PD را ارائه میدهد. هر دو نوع تغییر شکل غیر بیماری و بیماری برای ژندرمانی PD در مطالعات حیوانی و انسانی مورد آزمایش قرار گرفتهاند. درمانهای تغییر دهنده غیر بیماری که سنتز دوپامین یا GABA را هدف قرار میدهند، در بهبود علائم شناسی PD در مطالعات بالینی تصادفی موفق و امیدوار کننده بودهاند، اما آزمایش اساسی قبل از این که بتوان آنها را در مجموعه درمان بالینی استاندارد اجرا کرد، باقی ماندهاست. همانند اهداف تغییر بیماری که از لحاظ نظری امکان کند شدن پیشرفت بیماری را ارائه میدهند، چندین عامل نوروتروفیک نتایج دلگرمکنندهای را در مدلهای پیش بالینی نشان میدهند (به عنوان مثال، نورتورین، GDNF، BDNF، CDNF، VEGF - A). با این حال، تاکنون، آزمایشها بالینی تنها نورتورین را مورد آزمایش قرار دادهاند، و متاسفانه، هیچ آزمایشی قادر به رسیدن به نقطه پایانی اولیه آن نبوده است. آزمایشهای بالینی آینده با عوامل نوروتروفیک به وضوح شایسته انجام هستند، با در نظر گرفتن هدف فریبنده در واقع کند کردن روند بیماری PD. به عنوان انواع جایگزین ژندرمانی، اپتوو و کموژنتیک نیز ممکن است در آینده در درمان PD مورد استفاده قرار گیرند و تکنولوژی جدید ویرایش ژنوم نیز میتواند به طور بالقوه به عنوان ژندرمانی فردی برای انواع ژنتیکی PD به کار رود.
کلمات کلیدی: ژندرمانی، اهداف بیماری پارکینسون، دوپامین، GAD، عوامل نوروتروفیک، NRTN، CDNF، MANF، BDNF، GDNF، اپتوژنتیک، کموژنتیک، ویرایش ژنوم
مقدمه
بیماری پارکینسون (PD)دومین بیماری شایع نورودژنراتیو است و تخمین زده میشود تا ۵.۸ میلیون نفر در سراسر جهان مبتلا شوند [ ۱ ]، با افزایش سن متوسط جمعیت در جهان غرب، شیوع این بیماری در حال افزایش است. PD با شروع آهسته و نامتقارن علائم حرکتی به شکل سفتی عضلانی، اختلال در راه رفتن، هیپوکینزیا و لرزش و همچنین چندین علامت غیر حرکتی از جمله احساسات آشفته، شناخت، الگوی خواب و اختلال در عملکرد مستقل مشخص میشود [ ۲ ].
پاتوفیزیولوژی اساسی شامل تجمع سلولی سینوکلئین (SNCA)، تجمع در ساختارهای نامحلول است که منجر به اختلال عملکرد سلولی و سمیت میشود [ ۳ ]. این امر به ویژه با از دست دادن نورونهای دوپامینرژیک (DA)در بخش متراکم جسم سیاه (SNc)آشکار است که مسیرهای بسیاری را که وابسته به تونوس دوپامینرژیک ناشی از این جمعیت نورون هستند، مختل میکند. عدم تعادل منجر به سیگنالهای سرگردان در مدارهای سیستم جسم مخطط میشود که منجر به ظهور فنوتیپ پارکینسونین میشود [ ۲، ۴ ]. تجمع SNCA نیز یک مشخصه پاتولوژیک از گروهی از بیماریهای مغزی دیگر مانند جنون جسم لووی [ ۵ ] و آتروفی سیستم چندگانه [ ۶ ] است. این بررسی بر ژندرمانی برای PD تمرکز خواهد کرد، اما از آنجا که بسیاری از درمانهای پیشنهادی لزوما PD را هدف قرار نمیدهند، این احتمال وجود دارد که آنها بتوانند برای درمان synucleinopathies نیز عملی باشند.
رژیمهای درمانی فعلی برای PD عمدتا شامل تجویز لوودوپا (L - DOPA)، آگونیست های دوپامین یا مهار کنندههای MAO - B یا جراحی به شکل تحریک عمیق مغز (DBS)یا جراحی neuroablative است [ ۷، ۸ ].با این حال، این درمانها نشانهای هستند و از پیشرفت PD جلوگیری نمیکنند و ممکن است با عوارض جانبی قابلتوجهی همراه باشند. بنابراین، بررسی راههای درمانی جدید به وضوح تضمین شدهاست [ ۹ ].
ژندرمانی برای اولین بار در سال ۱۹۷۲ به عنوان وسیلهای برای "جایگزینی DNA بد با DNA خوب" توصیف شد [ ۱۰ ]. اصل اساسی هنوز هم پا بر جا است اما تا حدودی پیچیدهتر شدهاست. ژندرمانی میتواند برای درمان بیماریها با معرفی ژنهای درمانی و یا با جایگزینی، ساکت کردن و یا اصلاح ژنهای معیوب به کار رود. رویکردهای مختلفی وجود دارد، اما استراتژی اصلی استفاده از ناقل های ویروسی غیر تکراری مهندسی شده است؛ سروتایپ های مختلف ویروس نوترکیب آدنوویروس (AAV)یا لنتی ویروس [ ۱۱ ]. چندین مطالعه با استفاده از ناقل های ویروسی شواهدی را برای ایمنی و صحت بالای بیان ژن فراهم کردهاند [ ۱۲ - ۱۵ ]. روشهای جدید انتقال ژن در حال توسعه هستند و با موفقیت در مدلهای حیوانی PD مورد استفاده قرار گرفتهاند [ ۱۶، ۱۷ ]. در سالهای اخیر، ژندرمانی نیز در چندین آزمایش بالینی انسانی که در زیر بررسی شدهاند، مورد آزمایش قرار گرفتهاست. با تایید ناقل های AAV برای ژندرمانی در اروپا و اخیرا در ایالاتمتحده [ ۱۸، ۱۹ ]، انتظار میرود که رویکرد ژندرمانی زمینه بیشتری را در سالهای آینده به دست آورد. این بررسی یک نمای کلی به روز از ژندرمانی in vivo برای PD ارائه خواهد داد.
تاکنون چندین هدف ممکن برای درمان ژنتیکی PD شناسایی شدهاند. این اهداف را می توان به عنوان اصلاح بیماری یا اصلاح غیر بیماری دستهبندی کرد. درمانهای تغییر دهنده غیر بیماری با تلاش برای نرمال کردن شلیک نابجا در ganglia قاعدهای با بیان آنزیمهای دوپامینرژیک و یا GABAgenic هدف علائم اختلال حرکتی هستند. این درمانها علامتدار هستند و روند پاتوفیزیولوژیک اساسی را تغییر نمیدهند.
استراتژیهای تغییر بیماری حول توقف مرگ سلولی با واسطه PD و / یا تولید مجدد نورونهای از دست رفته میچرخد. بیشترین رویکرد مورد بررسی بیان بیش از حد عوامل رشد است که دارای خواص محافظ عصبی هستند (جدول ۴ و ۵ را ببینید)[ ۲۰ ]. سایر اهداف ممکن شامل ترمیم ژنومی ژنهای معیوب هستند [ ۲۱ ]، که منجر به ظهور ساختارهای سمی SNCA میشود.
اهداف اصلاحی غیربیماری زا
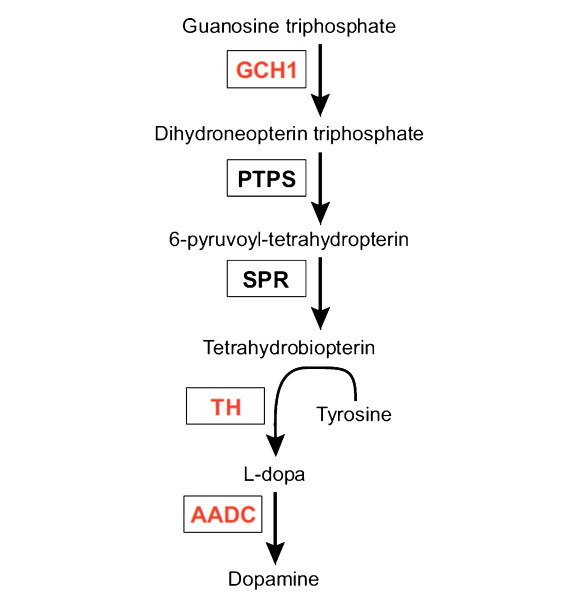
آنزیم دکربوکسیلاز اسید آروماتیک (AADC)بخشی از دستگاه سنتز دوپامین است (شکل ۱)و مسئول تبدیل L - DOPA به دوپامین است [ ۲۲ ]. رویکرد بالینی اولیه در مورد درمان PD، استفاده از L - DOPA است، یک پیشساز دوپامین که قادر به عبور از سد خونی - مغزی است، بنابراین درمان خوراکی را ممکن میسازد.
L- DOPA یک درمان علامتی است که قادر به توقف پیشرفت بیماری نیست و با چندین عارضه جانبی، از جمله به اصطلاح حالت خاموش، در ارتباط است.
حالات خاموش اغلب به علت عملکرد نامنظم دارو همراه با افسردگی و یا هیپومانیا میباشند.
این دورهها به تخلیه غیرقابلپیشبینی معده، بار بیش از حد سیستم حامل سد خونی مغزی که مسئول انتقال L - DOPA و همچنین ناکافی بودن سطح آل - آمینو اسید دکربوکسیلاز آروماتیک (AADC)است، نسبت داده میشود.
مسائل جذب تا حد زیادی با استفاده از فرمولهای جدید L - DOPA و سیستمهای پمپ مختلف برطرف شدهاست.
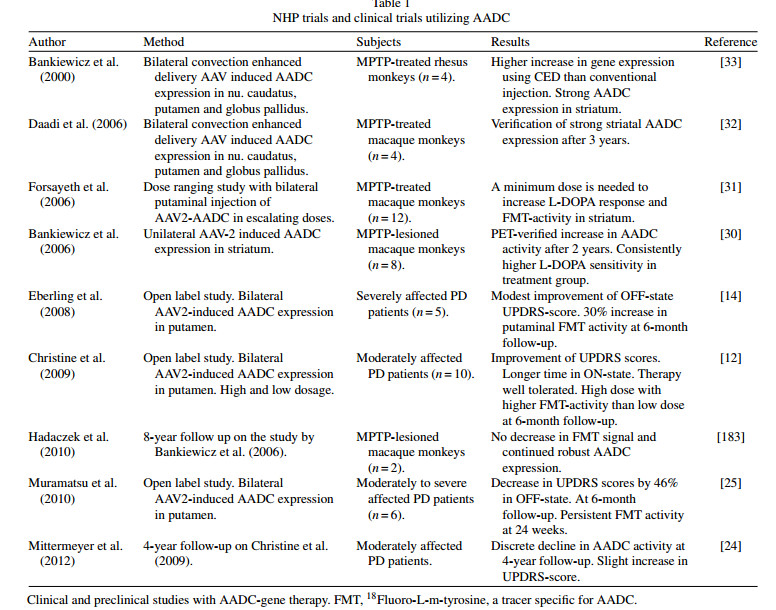
چندین مطالعه جوندگان نشان دادند که بیان بیش از حد AADC با واسطه جسم مخطط به خوبی تحمل میشود و قادر به بهبود علائم پارکینسون است [ ۲۷ - ۲۹ ]. مطالعات بیشتر در پریمات های غیر انسانی (NHP)[ ۳۰ - ۳۳ ] کاهش نیاز به دارو، و همچنین بهبود عملکرد حرکتی دارو را زمانی که سطح خاصی از ترانسفکشن به دست آمد، توصیف کرد (جدول ۱ را ببینید)[ ۳۱ ]. نتایج امیدوار کننده NHPs منجر به مرحله اول کارآزمایی بالینی با بیان AADC دو جانبه القا شده توسط AAV۲ در پوتامن بیماران PD به شدت تحتتاثیر که در سال ۲۰۰۸ منتشر شد، شد. نویسندگان بهبود اندکی در مقیاس یکپارچه درجهبندی بیماری پارکینسون (UPDRS)یافتند و از همه مهمتر، هیچ اثر زیان آوری از افزایش بیان AADC با واسطه AAV در انسان نیافتند. علاوه بر این، آزمایشهای فاز ۱ (جدول ۱ را ببینید)با توجه به علائم بهبود یافته در بیماران به نتایج مشابهی دست یافتند، در حالی که در حالت خاموش و همچنین کاهش کلی در UPDRS و گرایش به سمت نیاز کمتر به L - DOPA [ ۱۲، ۲۵ ]. یک مطالعه پیگیری بر روی بیماران یک مطالعه فاز ۱ توسط کریستین و همکارانش [ ۱۲ ] افزایش گسسته در امتیازات UPDRS و همچنین کاهش جزئی در فعالیت لیگاند AADC ۱۸ فلورو - L - تیروزین را نشان داد. با این حال، هر دوی آنها هنوز هم بالاتر از خط پایه بودند. نویسندگان این مقاله این کاهش را به تخریب مداوم نورونهای nigral که هنوز هم به کار خود ادامه میدهند نسبت میدهند، که به این معنی است که AAV درمانی بدون هیچ کاهشی در بیان سلولهای ترانسفکت شده، دائمی خواهد بود. در حال حاضر دو مطالعه در حال انجام و یک مطالعه با برچسب باز برنامهریزیشده با هدف بررسی پروفایل ایمنی درمان AADC، بهینهسازی دوز و روش تحویل وجود دارد [ ۳۴، ۳۵ ]. امیدوارم این مطالعات راه را برای تحقیقات تصادفی کنترلشده با دارونما هموار کنند.
اگر درمان AADC کارآمد به نظر برسد، به دلیل نیاز مداوم به L - DOPA بیرونی، به عنوان یک درمان مستقل مناسب نیست. با این حال،
با توجه به اینکه بیماران در تمام آزمایشها بالینی بهبود پاسخ L - DOPA و همچنین بهبود کلی نمرات UPDRS را گزارش کردهاند، این احتمال وجود دارد که ژندرمانی AADC بتواند در درمان آینده با هدف بهبود نوسانات حرکتی غیرقابل کنترل دیگر گنجانده شود و با DBS PD در درمان مورد استفاده قرار گیرد.
شکل ۱. آل - تیروزین توسط تیروزین هیدروکسیلاز (TH)با استفاده از عامل مشترک تتراهیدروبیوپترین، تحت حد سرعت GTP سیکلوهیدروکسی - لاز ۱ (GCH۱)به آل - دی هیدروکسی فنیلآلانین (L - DOPA)تبدیل میشود. L- DOPA بعدا توسط آلفا آمینو اسید دکربوکسیلاز (AADC)به دوپامین تبدیل میشود. سه آنزیم قرمز توسط ژندرمانی پروساوین کد گذاری میشوند. PTPS: pyruvoyltetrahydropterin synthase؛ جدول ۱: آزمایشهای NHP و کارآزمایی بالینی با استفاده از AADC
جدول ۱: آزمایشهای NHP و کارآزمایی بالینی با استفاده از AADC
AADC، TH و GCH
یک رویکرد فریبنده برای درمان علامتی PD، بازسازی کامل دستگاه سنتز دوپامین با معرفی تمام ژنهای مسئول تبدیل تیروزین به دوپامین (شکل ۱)در سلولهای منطقه هدف است. این امر نیازمند آنزیم تیروزین هیدروکسیلاز (TH)همراه با عامل مشترک تتراهیدروبیوپترین (BH۴)برای تبدیل آمینو اسید آل - تیروزین به L - DOPA است که به نوبه خود توسط AADC به دوپامین تبدیل میشود. مرحله محدود کننده سرعت برای سنتز BH۴ آنزیم GTP سیکلوهیدروکسیلاز ۱ (GCH)است [ ۳۶ ]. با بیان سه آنزیم (TH، GCH و AADC)در جسم مخطط، باید از لحاظ نظری امکان کاهش، اگر حذف نشود، نیاز به L - DOPA بیرونی، که میتواند یک تونوس دوپامین پایدار را تضمین کند که اثرات از بین رفتن و اثرات خارج از هدف را از بین میبرد، وجود داشته باشد.
یک مطالعه اولیه توسط کپلایت و همکاران (۱۹۹۴)بهبود رفتاری را در موشهای صحرایی تیمار شده با ۶ - OHDA که در جسم مخطط در معرض AAV - TH قرار گرفتند، گزارش کرد. این روش در یک مطالعه NHP توسط همکاران (۱۹۹۸)که ترکیبی از AAV - TH و AAV - AADC را در جسم مخطط میمونهای سبز تزریق کردند، مورد بررسی قرار گرفت. با این حال، آنها قادر به تشخیص هیچ گونه اثر رفتاری درمان در مقایسه با کنترلهای تزریقشده با وکتور خالی نبودند. با این وجود، آنها توانستند افزایش تولید دوپامین در جسم مخطط را تشخیص دهند. فقدان اثر بالینی در مطالعه آنها احتمالا میتواند به دلیل بازیابی تحت بالینی سطح دوپامین به دلیل مقادیر کم وکتور تزریقشده [ ۳۸ ] یا فقدان BH۴ باشد.
بعدا، بیشتر تحقیقات در مورد بازسازی دستگاه دوپامینرژیک بر بیان هر سه آنزیم دوپامینرژیک متمرکز شدهاست. با این حال، انتقال هر سه ژن یک مانع را نشان میدهد، چرا که سه آنزیم ترکیبشده بسیار بزرگ هستند و نمیتوانند در یک ژنوم AAV قرار گیرند. این چالش به دو روش حل شدهاست. یا با تزریق مخلوطی از سه vectors AAV متفاوت، که هر کدام حاوی یکی از ترانسژن های مورد نظر هستند، یا با استفاده از LV، که ظرفیت بیان ترانسژن بالاتری دارد. مطالعات جوندگان نشان میدهد که هر دو روش به خوبی تحمل میشوند و در کاهش فنوتیپ پارکینسونین در مدلهای PD کارآمد هستند [ ۳۹ - ۴۷ ].
این موفقیت باعث تحقیقات بیشتر در NH ها (جدول ۲ را ببینید)با هر دو رویکرد تریسیستونیک و لنتی ویرال شد. یک مطالعه اولیه با استفاده از بردارهای تریسستونیک نشان داد که افزایش بیان TH، AADC و GCH [ ۴۸ ] هم ممکن است و هم به خوبی تحمل میشود. با در نظر گرفتن عدم وجود اثر بالینی مشاهدهشده در مطالعه توسط در طول و همکاران (۱۹۹۸)، مورتامسو و همکاران (۲۰۱۰)یک تیتر افزایشیافته را تزریق کردند و AAV۲ - GCH را به تزریقات اضافه کردند و یک افزایش قابلتوجه در تولید دوپامین و همچنین یک نجات قوی از فنوتیپ پارکینسونین در موشهای صحرایی تیمار شده با MPTP [ ۴۸ ]، با اثر تداوم در پیگیری ۱۵ ساله در یک میمون [ ۴۹ ] ارائه دادند.
پیشنهاد شدهاست که بستهبندی همه ژنها در یک وکتور، بیان مشترک پروتئینها را تضمین میکند و در نتیجه مانع از مشکل سیستم تریسستونیک میشود. نشانداده شدهاست که رویکرد LV در موشها کارآمد است [ ۳۹ ]. ارائه یک مطالعه بر روی اثرات یک وکتور LV بیانکننده AADC، TH و GCH در پوتامن میمونهای مکاکی تیمار شده با MPTP انجام شده است. این روش درمانی به خوبی تحمل شد، و افراد مورد مطالعه بهبود قابلتوجه علائم پارکینسون را بدون هیچ نشانهای از دیس کینزی نشان دادند. میکرودیالیز ترمیم تقریبا ۵۰ درصدی تونوس دوپامین نرمال را نشان داد [ ۵۰ ]. یک آزمایش فاز ۱ بعدی که اثر تزریق LV - TH - GCH - AADC تحت نام پروساوین به پوتامن را بررسی میکرد، توسط شرکت آکسفورد بیومدیکا حمایت مالی شد [ ۵۱ ]. نویسندگان این مطالعه دریافتند که این دارو به خوبی تحمل میشود و قادر به کاهش نمرات UPDRS در بیماران بدون دارو تا ۱۲ امتیاز و همچنین کاهش نیاز عمومی به دارو میباشد. این مطالعه با چندین بازدید برنامهریزیشده برای ارزیابی بیشتر ایمنی و کارایی طولانیمدت ادامه دارد [ ۵۲ ]. مکانیزم پشت اثر ظاهری پروساوین هنوز به طور کامل درک نشده است، اما این نظریه مطرح شدهاست که افزایش سطح دوپامین در محیط خارج سلولی که توسط سلولهای ترانسفکت شده به دست میآید، میتواند نرخ آتش محورهای افکنش آوران را به همان شیوه DBS نرمال کند [ ۳۸، ۵۱، ۵۳ ]. بنابراین، تنها کارآزمایی بالینی در پروساوین، از تحقیقات بیشتر در مورد دوز بردار قبل از اینکه امکان پیشرفت به مطالعه فاز ۲ وجود داشته باشد، حمایت میکند [ ۵۱ ].
انتقالدهنده مونوآمین وزیکولار ۲ (VMAT۲)یک ماکرومولکول است که دوپامین را به وزیکول های سیناپسی داخل سلولی نورونهای دوپامینرژیک برای آزاد شدن در شکاف سیناپسی منتقل میکند [ ۲۶ ].
ادامه دارد (البته احتمالا)
برچسب های مهم
مطالب تصادفی






/cloudfront-us-east-2.images.arcpublishing.com/reuters/IE22OFLXBRLWZFPBRQMDYEXZVE.jpg)
